




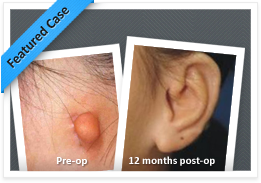
PATIENT RESULTS
Atresia
Surgery to repair congenital aural atresia (CAA) is one of the most challenging operations the otology specialist faces. The goals of surgery are to provide the patient with a clean, skin-lined external auditory canal with long-term restoration or improvement in hearing.
In congenital aural atresia, the external auditory canal (EAC) and structures in the middle ear fail to develop completely. Development of the ear canal and middle ear may be arrested at any point in the process. Therefore, the clinician may encounter varying degrees of severity of this malformation. In the severe form of the disorder, no identifiable ear canal exists (complete atresia), and the middle ear and its structures (ossicles, or ear bones) may be absent or show significant underdevelopment. If some sort of external auditory meatus is present, the ear canal may end in a shallow blind pouch. In less severe forms of the disorder, the ear canal may be stenotic with a pinpoint aperture leading into the medial ear canal and, possibly, a rudimentary tympanic membrane. The tympanic membrane in these cases may or may not be attached to the ear bones (the ossicular chain).
Congenital aural atresia is commonly accompanied by microtia.
High resolution computed tomography (CT) scanning has allowed the otologist to “see” into the middle and inner ears to understand the internal anatomy. Before CT scanning, plain radiography and tomography were available, and surgeons could only guess what the anatomy looked like inside without the ability to predict good surgical candidates.
In CAA, the role of the otologist extends beyond diagnosis and treatment. The otologist must consider the hearing needs of the child, especially with regard to bilateral atresia, and must determine not only the appropriate treatment, but also the timing of that treatment. The otologist must work closely with the plastic surgeon to determine the optimal time for atresia repair in relation to the microtia repair.
Not all patients with CAA are candidates for surgical correction. Among patients with associated syndromes such as Treacher Collins or Hemifacial microsomia (HFM)/Goldenhar, approximately 50% of patients are not surgical candidates because of the existing anatomy. In isolated unilateral cases of CAA (most patients) approximately 65-75% will be surgical candidates however a significant number will not feel they need the surgery.
For surgical candidates with favorable anatomy, the otologist must rely on the skills, techniques, and experience from middle ear and mastoid surgery to optimize the surgical result. Surgical success is based on restoration of useful hearing, long-term stability of hearing, and maintenance of a patent, skin-lined ear canal.
With a few minor modifications, the technique of Jahrsdoerfer first described in 1973 is still used today and continues to deliver significantly improved hearing results in carefully selected patients.
The Problem
The main anatomic deformity in CAA is failure of the external ear canal to complete development. The severity of the deformity is variable. In the most severe form of CAA (ie, when development of the ear is halted early), the ear canal cannot be identified. Bone fills the region the ear canal normally occupies, and no external opening (meatus) is present. In mild cases, the ear canal is present, but it is stenotic and markedly narrowed. A rudimentary tympanic membrane may be present, and it may or may not be connected to the ossicular chain.
Most patients with CAA have normal inner ear (cochlear) function since it develops earlier and from a different structure.. The problem is simply that the sound energy is not being transmitted or conducted to the inner ear. Ways to improve the transmission of sound to the healthy inner ear include the bone conducting hearing aid, the BAHA system, and surgery to open the ear canal and restore the natural sound conducting mechanism to the inner ear (atresia repair or atresia surgery).
What causes CAA ?
Factors that cause the developmental sequence of the ear canal and middle ear to cease in CAA are not known. Regions adjacent to the ear canal, including the jaw bone (mandible) may be affected as well.
CAA commonly coexists with microtia. However, rarely, CAA may occur alone with an auricle that appears healthy. The embryologic precursors of the auricle/external ear are called hillocks. Each of the 6 hillocks is responsible for forming a distinctive part of the auricle.The auricle assumes adult shape (but not size) by the 20th week of fetal life. Microtia (“small ear”) results when the development of these hillocks is arrested. In general, the middle ear is less developed when microtia is severe than when it is not but this is not always the case and the opposite may be true.
Types of CAA
Colman graded the severity of CAA into 3 categories: minor, moderate, and severe. Patients with severe CAA have total canal atresia and usually present with unfavorable middle ear anatomy and temporal bone development for reconstruction. In comparison, patients with moderate CAA have more favorable anatomy (ie, an identifiable ear canal with deformed ossicles and an aerated middle ear space). Most patients with CAA present with this moderate form. In minor cases, the ear canal is present but narrowed, and the middle ear is better developed.
CAA may coexist with syndromes that feature first and second branchial arch deformities (eg, Treacher-Collins syndrome, hemifacial microsomia, Goldenhar syndrome, and other craniofacial abnormalities).
Detection at birth
If the anatomic abnormality is detected at birth, the otologist meets the newborn and parents in consultation. The parents are usually anxious and eager to have their many questions answered. Reassurance and parent education are critical during this time. Regardless of whether this condition is present in 1 or both ears, cochlear function must be assessed in both ears by means of specialized audiologic testing, such as auditory-evoked brainstem responses (ABR or BAER testing) elicited from both air-conducted and bone-conducted signals. If bilateral CAA and good cochlear function are present, the infant needs amplification with a bone-conducting hearing aid (eg, BAHA Softband) until a decision is made to pursue surgical correction just before the child starts school.
Amplification has traditionally not been recommended in children with unilateral CAA as long as the contralateral ear functions well. In general, normal hearing in one ear is enough hearing to support normal speech and language development. Reliable data to the contrary is yet to appear. However I encourage all parents of a newborn child with unilateral microtia/atresia to try a hearing aid of some sort from early in life . Sometimes a child will wear one and continue to do do while others will wear an aid in the first 18 months , only to pull it off as a two/three year old despite the wishes of parents. We can only try ! We do not know how much benefit is gained by this early aiding but we can only assume there may be some ? Parents should be reassured if the aiding does not work for them that both sides of the brain are stimulated by the one good ear so anatomy is on our side !
Canal stenosis and the risk of cholesteatoma
A child with canal stenosis (mild atresia) may present to the clinician with or without associated hearing loss in the ear. These patients with narrowed canals (generally less than 2 mm are at risk of canal cholesteatoma. Cholesteatoma is a pocket or cyst of skin that grows and expands as dead skin cells fill the cyst. Careful observation, follow-up, and microscopic examination and cleaning may ensure the patient does not develop a canal cholesteatoma. However, when the opening to the ear canal is too small to permit examination or when the patient’s history ( discharge, ear pain) suggests a cholesteatoma, radiographic imaging with CT scanning is used to evaluate the possibility of a canal cholesteatoma .
Indications
Before surgical correction is considered, the patient must fulfill 2 criteria. First, the patient with congenital aural atresia (CAA) must have normal cochlear function, as demonstrated on audiologic examination and normal bone conduction thresholds in the atretic ear. Second, CT scanning must demonstrate normal inner ear and internal auditory canal morphology.
If patients meet these criteria, the next step is to identify patients with favorable anatomy who have a reasonable chance of success with surgical correction. Jahrsdoerfer devised a 10-point grading scheme, based on features from the CT scan and appearance of the external ear, that reflects the likelihood of success .Patients who have a score of 7 or higher are good candidates for surgery. Patients must also be motivated to undergo the surgery and postoperative care and must be willing to have the ear cleaned of dead skin once or twice a year for the rest of their lives. In addition the outer ear reconstruction is better to be done first and whether it is rib or Medpor a canal reconstruction can detract from the appearance of a good ear reconstruction by appearing as a large “hole” in the middle of an otherwise good looking ear and for some patients this will be an issue .
Alternatives to surgical correction include amplification devices, such as a bone-conducting hearing aid or bone-anchored hearing device/BAHA system Conventional hearing aids in patients with canal stenosis may also be used if the hearing device is able to provide adequate amplification. Distinction should be made between the Baha system (which is the osseointegrated titanium implant and speech processor, and the Baha Softband which is a bone conducting hearing device that is worn on a soft headband around the head. The Baha system is FDA-approved for children 5 and older in the United States.
Experience using the BAHA device is considerable, especially in Europe. Long-term results of the US experience with the BAHA are also favorable. This device features a surgically implanted, percutaneous titanium fixture-abutment that osseointegrates into the temporal bone. A sound transducer attaches to this titanium fixture and delivers the sound energy directly to the cochlea via bone conduction. Therefore, one does not need an ear canal or middle ear to hear.
This type of technology is more efficient than the head band or soft band bone-conducting hearing aids because the sound energy is not attenuated by the skin and intervening soft tissues. Speech reception thresholds (SRTs) less than 30 dB are obtainable with the use of these devices. Bilateral BAHA devices have even been used for patients with bilateral atresia. Bilateral implants may impart better sound localization ability and possibly better hearing in noisy environments. Furthermore, the use of the BAHA device does not preclude future reconstructive surgery. Potential complications of BAHA placement include failure of osseointegration with extrusion of the fixture/abutment, skin overgrowth over the abutment, and flap necrosis with secondary healing.
These alternatives may be considered in patients with unilateral or bilateral congenital aural atresia (CAA) who do not desire surgical correction or in whom the existing anatomy is not favorable (a score of 6 or less on the Jahrsdoerfer CT grading scale) but the cochlea is still functional and can be stimulated.
Timing of surgery
External ear reconstruction is usually delayed until the child is about 7-10 years or as requested by the patient, with atresia repair following the series of operations required for rib graft microtia repair. This delay allows for growth of the rib cage, enabling sufficient costal cartilage to be harvested for sculpting the auricle. A child who is large for his/her age may be operated on earlier. The atresia surgery is typically performed after the cartilaginous auricular framework is placed.
Although reconstructive surgeons using the Medpor implant have implanted children as young as 3 years, we do not advocate atresia repair at that age because the child is simply too young to cooperate with the necessary postoperative packing removal and ear canal cleaning which is so critical to the success of the operation long term. In addition, younger children may have a higher rate of meatal or canal stenosis necessitating revision surgery. We have never seen a child bothered psychologically by the ear deformity before the age of 5 years let alone interested in any surgery for microtia. Even after 5 years of age teasing and psychological issues have rarely arisen and children in our experience continue to resist the idea of surgery unitl pre-teen and teenage years in many casesThe benefits of binaural hearing and improved sound localization must be weighed against the potential complications of surgery, including canal stenosis and new bone growth seen in younger patients. Bearing in mind in patients after the age of 7 years in large published series the need for revision of canal surgery to re-open a canal that has closed is reported at between 10-25% and knowing from our experience in craniofacial patients that infants skulls form new bone vigorously it seems likely that the revision rate in childen less than 5 years old would be even higher. In addition if atresia is preformed at the sam time as or after a Medpor reconstruction there is a chance with further surgery that the Mepor implant could be partly exposed and at risk.
Surgery in patients with bilateral CAA ( if anatomically feasible) is performed with the goal to restore useful hearing, unaided or aided. Surgery should take place just before the child enters school, usually when aged 5-6 years.
Complications
Injury to the facial nerve is one of the most feared complications of surgery for correction of congenital aural atresia (CAA). This complication has historically deterred surgeons from correcting this condition. However, in experienced hands and with improved imaging techniques, the complication rate has been below 1%.
The facial nerve is estimated to be aberrant ( in an abnormal location ) in 25-30% of patients so this possibility should be anticipated and a facial nerve monitor used to warn the otology surgeon when they are getting too close to the nerve.
Complications of atresia surgery can be minimized with proper selection of patients and with careful attention to surgical detail. Most common complications include meatal or canal re-stenosis in 15-20% (even in experience hands), usually requiring revision surgery, and chronic drainage/infection in 10%, usually due to shedding of the the skin graft with “mucosalization” of the canal, which requires revision with a new skin graft . Less commonly, sensorineural hearing loss (5%) is most likely related to the high energy of the drill on the ossicular chain being conducted to the cochlea, and facial nerve injury occurs in 0.1%. About 15-20% of patients also lose the initial gains in hearing, due either to lateralization of the tympanic membrane ( shifting of the new ear drum outwards and out of contact with the free ear bones/ossicle) or refixation’fusion of the unfused ossicular chain.
Outcome and Prognosis
The goal of congenital aural atresia (CAA) surgery is to restore hearing to a speech-reception threshold (SRT) of less than or equal to 30 dB HL without the need for amplification. The 2 most important factors in achieving consistently good hearing outcomes in atresia surgery are careful preoperative selection of patients and meticulous surgical technique at each step of the operation. Surgery is not recommended for patients with unilateral atresia and Jahrsdoerfer scores of 6 or below. Surgery is attempted on the patient with bilateral atresia and a score of 5 or 6, but excellent hearing outcomes are difficult. Even so, the patient may be able to wear a conventional hearing aid in the new canal to bring the hearing thresholds into the normal range. Surgery is not recommended for patients with 4 or below. The most important anatomic feature for successful surgery is middle ear aeration; without an aerated middle ear space, the patient is not considered a candidate for surgery.
Some patients with atresia may require reconstruction of the ear bones with an artificial ear bone, or prosthesis if a vital bone is not present. This type of reconstruction tends not to be as reliable as using the patient’s own native ossicular chain. Anatomy of the middle ear and ossicles occasionally dictates (approximately 8-10% of the time) that a prosthesis must be used.
Future and Controversies
Improvement in the ability to treat congenital aural atresia (CAA) lies with ways to maintain patency of the surgically reconstructed ear canal. This improvement may come in the form of genetically engineered tissue grafts or advanced stent materials.
Is there an age that is too young to repair CAA? Certainly the child who cannot sit still in the office for the necessary packing removal and ear canal cleaning is not old enough to undergo the surgeryIn addition a useful surgically made ear canal needs to be made about 1.5cm in diameter . It is hard enough in the near adult size skulls of patients older than 6 years to negotiate the vital structures in this region and still have space for such a large canal let alone in a much young ,smaller skull which may allow even less space to safely work between these vital areas. In addition a widely patent surgically made ear canal in the middle of an ear reconstruction can look unusually large from an aesthetic point of view and may bother the patient.
Key Points: the controversy of surgery for unilateral atresia
The auditory system, unlike the visual system, sends signals from both ears to both sides of the brain. Central auditory pathways cross the midline very early in the auditory pathway so that both sides of the brain receive stimulation from both ears. This means that both sides of the brain are being stimulated by the one good ear in unilateral aural atresia. The argument that the earlier the surgeon opens the atretic ear, the less chance the brain will “close down” (and not be able to respond) to input from the reconstructed ear is completely spurious and misleading. Thus patients will enjoy the same benefits of this surgery whether they have atresia surgery at 3 or 30 years of age! Also misleading is the claim that unilateral atresia patients will fail more at school and have reduced acedemic or even earning potential ! These children mostly have normal academic potential with or without atresia correction most of our patients are achieving academic milestones or exceeding them. However is is recommended that all newborn children born with unilateral aural atresia trial a bone conduction hearing aid on the assumption that there may be some benefit gained in terms of amplication early on. Parents should ask at an Australian Hearing Centre to trial free of charge a “Compact Mini “ device which is similar to a BAHA soft band hearing aid. The benefit is not proven as yet but we all assume to some degree that two ears are better than one. In the future evidence may emerge to refute or support the use of these devices. However parents should not get frustrated if they find their 1 year old uses the device only to later pull it off in frustration as a two or three year old ! Sometimes children just have their own ideas about wearing things on their heads !
Unilateral atresia patients mostly complain that their sound localization is poor not so much their overall hearing. It is important to remember that excellent studies have shown that in unilateral atresia patients having atresia corrected that sound localization and some tasks of hearing in noisy environments remain elusive for many postoperative atresia patients, even with excellent hearing outcomes (1,2,3,4).
The difficulty patients have with one good ear and one atretic ear is difficulty hearing in background noise and difficulty locating a sound source. When successful surgery to open the atretic ear canal and restore hearing to the ear will eliminate (or greatly reduce) these problems, whether the patient is 3 or 30! In fact work by Brad Kesse and his group actually suggests that teenagers having atresia surgery may do better than both younger children or older adults in terms of actually learning to use their new hearing after an atresia procedure.
Another really important point is that atresia surgery should be undertaken by a atresia surgeon who is in your local area. Long term a minimum of 15-20 % of patients having a successful atresia procedure experience deterioration in their result for a number of reasons and need a revision procedure. In fact in the most experienced groups in the world the rates of revision are 15-58% ! For this reason the procedure is best undertaken by the surgeon who can continue caring for the new ear canal long term. Unlike microtia surgery for the outer ear shape , atresia surgery requires long term maintenance.
It must be remembered that some activities such as scuba diving or serious ocean sports may not be possible after atresia surgery and if your child is likely to want to be involved in these later on it may be a good argument to wait until teenage years to be able to consider this aspect and the implications before committing to atresia surgery.
References
1. . Lambert PR. Congenital aural atresia: stability of surgical results. Laryngoscope. Dec 1998;108(12):1801-5.
2. De la Cruz A, Teufert KB. Congenital aural atresia surgery: long-term results. Otolaryngol Head Neck Surg. Jul 2003;129(1):121-7
3. Gray L, Kesser BW, and Cole EA. Understanding speech in noise after correction of congenital unilateral aural atresia: effects of age on the emergence of binaural squelch but not in use of head-shadow. International Journal of Pediatric Otorhinolaryngology. 2009;73:1281-7
4. Wilmington D. Gray L. Jahrsdoerfer R. Binaural processing after corrected congenital unilateral conductive hearing loss. Hearing Research. April 1994;74:99-114.
OUR LOCATION
REQUEST AN APPOINTEMENT
After submitting the form above, you will be contacted by our staff to answer any questions and discuss scheduling an appointment. You can also call us directly at +61 3 9508 9508 or Lee Dowling PA to Dr Greensmith on leed@melbplastsurg.com.